


 216.73.216.16
216.73.216.16 User Stats:
User Stats:
 Today: 0
Today: 0 Yesterday: 0
Yesterday: 0 This Month: 0
This Month: 0 This Year: 0
This Year: 0 Total Users: 117
Total Users: 117 New Members:
New Members:
 216.73.xxx.xx
216.73.xxx.xx
 Server Time:
Server Time:
 |
|
 |
|
|
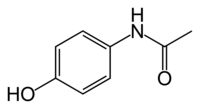
Paracetamol
|
|
| Systematic (IUPAC) name | |
| N-(4-hydroxyphenyl)acetamide | |
| Identifiers | |
| CAS number | 103-90-2 |
| ATC code | N02BE01 |
| PubChem | 1983 |
| DrugBank | APRD00252 |
| Chemical data | |
| Formula | C8H9NO2 |
| Mol. weight | 151.17 |
| Physical data | |
| Density | 1.263 g/cm³ |
| Melt. point | 169 °C (336 °F) |
| Solubility in water | 1.4g/100ml or .014 mg/mL (20 °C) |
| Pharmacokinetic data | |
| Bioavailability | almost 100% |
| Metabolism | 90 to 95% hepatic |
| Half life | 14 hours |
| Excretion | renal |
| Therapeutic considerations | |
| Pregnancy cat. | A(AU) B(US) |
| Legal status | S2(AU) GSL(UK) OTC(US) |
| Routes | oral, rectal, IV |
Paracetamol (INN) (IPA: [pærəˈsitəmɒl, -moʊl, -ˈsɛtə-]) or acetaminophen (USAN), is a common analgesic and antipyretic drug that is used for the relief of fever, headaches, and other minor aches and pains. Paracetamol is also useful in managing more severe pain, allowing lower dosages of additional non-steroidal anti-inflammatory drugs (NSAIDs) or opioid analgesics to be used, thereby minimizing overall side-effects. It is a major ingredient in numerous cold and flu medications, as well as many prescription analgesics. It is remarkably safe for human use in recommended doses, but because of its wide availability, deliberate or accidental overdoses are fairly common.
Common brand names for the drug include Herron in Australia, Tylenol in Brazil, Canada, South Korea and the U.S., Paralen in the Czech Republic and Slovakia, Panadol in Central America, Australia, New Zealand, Pakistan, Malaysia, Hong Kong, the UK, Portugal and Singapore, Perdolan in Belgium, Doliprane, Dafalgan, and Efferalgan in France, Tachipirina and Efferalgan in Italy, Crocin in India, Gelocatil in Spain, Benuron in Portugal, Alvedon in Sweden, Panodil and Pinex in Denmark and Iceland, Acamol in Israel, Pinex and Paracet in Norway, Depon in Greece and Lekadol in Slovenia.
The words acetaminophen and paracetamol both come from the chemical names for the compound: N-acetyl-para-aminophenol and para-acetyl-amino-phenol. In some contexts, it is shortened to apap, for N-acetyl-para-aminophenol.
Contents |
History
In ancient and medieval times, the only antipyretic agents known were compounds contained in white willow bark (a family of chemicals known as salicins, which led to the development of aspirin), and compounds contained in cinchona bark. Cinchona bark was also used to create the anti-malaria drug quinine. Quinine itself also has antipyretic effects. Efforts to refine and isolate salicin and salicylic acid took place throughout the middle- and late-19th century, and was accomplished by Bayer chemist Felix Hoffman (this was also done by French chemist Charles Gergardt 40 years earlier, but he abandoned the work after deciding it was too impractical).[1]
When the cinchona tree became scarce in the 1880s, people began to look for alternatives. Two alternative antipyretic agents were developed in the 1880s: Acetanilide in 1886 and Phenacetin in 1887. By this time, paracetamol had already been synthesized by Harmon Northrop Morse via the reduction of p-nitrophenol with tin in glacial acetic acid. While this was first performed in 1873, paracetamol was not used medically for another two decades. In 1893, paracetamol was discovered in the urine of individuals who had taken phenacetin, and was concentrated into a white, crystalline compound with a bitter taste. In 1899, paracetamol was found to be a metabolite of acetanilide. This discovery was largely ignored at the time.
In 1946, the Institute for the Study of Analgesic and Sedative Drugs awarded a grant to the New York City Department of Health to study the problems associated with analgesic agents. Bernard Brodie and Julius Axelrod were assigned to investigate why non-aspirin agents were associated with the development of methemoglobinemia, a condition that decreases the oxygen-carrying capacity of blood and is potentially lethal. In 1948, Brodie and Axelrod linked the use of acetanilide with methemoglobinemia and determined that the analgesic effect of acetanilide was due to its active metabolite paracetamol. They advocated the use of paracetamol (acetaminophen), since it did not have the toxic effects of acetanilide.[1]
The product went on sale in the United States in 1955 under the brand name Tylenol.
In 1956, 500 mg tablets of paracetamol went on sale in the United Kingdom under the trade name Panadol, produced by Frederick Stearns & Co, a subsidiary of Sterling Drug Inc. Panadol was originally available only by prescription, for the relief of pain and fever, and was advertised as being "gentle to the stomach," since other analgesic agents of the time contained aspirin, a known stomach irritant. In June 1958 a children's formulation, Panadol Elixir, was released.
In 1963, paracetamol was added to the British Pharmacopoeia, and has gained popularity since then as an analgesic agent with few side-effects and little interaction with other pharmaceutical agents.
The U.S. patent on paracetamol has expired and generic versions of the drug are widely available under the Drug Price Competition and Patent Term Restoration Act of 1984, although certain Tylenol preparations are protected until 2007. U.S. patent 6,126,967 filed September 3, 1998 was granted for "Extended release acetaminophen particles."
Available forms
Panadol, which is marketed in Europe, Asia, Central America, and Australasia, is the most widely available brand, sold in over 80 countries. In North America, paracetamol is sold in generic form (usually labelled as acetaminophen) or under a number of trade names: for instance Tylenol (McNeil-PPC, Inc), Anacin-3, Tempra, and Datril.
In some formulations paracetamol is combined with the opioid codeine, sometimes referred to as co-codamol (BAN). In the United States and Canada, this is marketed under the name of Tylenol #1/2/3/4 and in the U.S. is only available by prescription, while the lowest strength is over-the-counter in Canada. In the UK and in many other countries, this combination is marketed under the names of Tylex CD and Panadeine. Other names include Captin, Disprol, Dymadon, Fensum, Hedex, Mexalen, Nofedol, Paralen, Pediapirin, Perfalgan, and Solpadeine. Paracetamol is also combined with other opioids such as dihydrocodeine, referred to as co-dydramol (BAN), oxycodone or hydrocodone, marketed in the U.S. as Percocet and Vicodin, respectively. Another very commonly used analgesic combination includes acetaminophen in combination with propoxyphene napsylate, sold under the brand name Darvocet. A combination of paracetamol, codeine, and the calmative doxylamine succinate is marketed as Syndol or Mersyndol.
Acetaminophen is commonly used in multi-ingredient preparations for migraine headache, typically including butalbital and acetaminophen with or without caffeine, and sometimes containing codeine. In the U.S., these anti-migraine products are marketed under the brand name Fioricet.

It is commonly administered in tablet, liquid suspension, suppository or intravenous form. The common adult dose is 500 mg to 1000 mg. The recommended maximum daily dose, for adults, is 4 grams. In recommended doses paracetamol is safe for children and infants as well as for adults.
The effectiveness of paracetamol is often underestimated because of its widespread availability.
Mechanism of action
Paracetamol has long been suspected of having a similar mechanism of action to aspirin because of the similarity in structure. That is, it has been assumed that paracetamol acts by reducing production of prostaglandins, which are involved in the pain and fever processes, by inhibiting the cyclooxygenase (COX) enzyme.
However, there are important differences between the effects of aspirin and those of paracetamol. Prostaglandins participate in the inflammatory response which is why it has been known to trigger symptoms in asthmatics, but paracetamol has no appreciable anti-inflammatory action and hence does not have this side-effect. Furthermore, COX also produces thromboxanes, which aid in blood clotting aspirin reduces blood clotting, but paracetamol does not. Finally, aspirin and the other NSAIDs commonly have detrimental effects on the stomach lining, where prostaglandins serve a protective role, but paracetamol is safe.
Indeed, while aspirin acts as an irreversible inhibitor of
COX and directly blocks the enzyme's active site,
paracetamol indirectly blocks COX, and that this blockade is
ineffective in the presence of peroxides.[2] This might
explain why paracetamol is effective in the central nervous
system and in endothelial cells but not in platelets and
immune cells which have high levels of peroxides.
In 2002 it was reported that paracetamol selectively blocks a variant of the COX enzyme that was different from the then known variants COX-1 and COX-2.[3] This enzyme, which is only expressed in the brain and the spinal cord, is now referred to as COX-3. Its exact mechanism of action is still poorly understood, but future research may provide further insight into how it works.
A single study has shown that administration of paracetamol increases the bioavailability of serotonin (5-HT) in rats,[4] but the mechanism is unknown and untested in humans.
Metabolism
Paracetamol is metabolized primarily in the liver, where most of it (60-90% of a therapeutic dose) is converted to inactive compounds by conjugation with sulfate and glucuronide, and then excreted by the kidneys. Only a small portion (5-10% of a therapeutic dose) is metabolized via the hepatic cytochrome P450 enzyme system (specifically CYP2E1); the toxic effects of paracetamol are due to a minor alkylating metabolite (N-acetyl-p-benzo-quinone imine, abbreviated as NAPQI) that is produced through this enzyme, not paracetamol itself or any of the major metabolites. The metabolism of paracetamol is an excellent example of toxication, because the metabolite NAPQI is primarily responsible for toxicity rather than paracetamol itself.
At usual doses, the toxic metabolite NAPQI is quickly detoxified by combining irreversibly with the sulfhydryl groups of glutathione to produce a non-toxic conjugate that is eventually excreted by the kidneys.
Comparison with NSAIDs
Paracetamol, unlike other common analgesics such as aspirin and ibuprofen, has no anti-inflammatory properties, and so it is not a member of the class of drugs known as non-steroidal anti-inflammatory drugs (NSAIDs). In recommended doses, paracetamol does not irritate the lining of the stomach, affect blood coagulation as much as NSAIDs, or affect function of the kidneys.
Paracetamol is safe in pregnancy, and does not affect the closure of the fetal ductus arteriosus (as NSAIDs can). Unlike aspirin, it is safe in children as paracetamol is not associated with a risk of Reye's syndrome in children with viral illnesses.
Like NSAIDs and unlike opioid analgesics, paracetamol has not been found to cause euphoria or alter mood in any way. Paracetamol and NSAIDs have the benefit of bearing a low risk of addiction, dependence, tolerance and withdrawal.
Paracetamol, particularly in combination with weak opioids, is more likely than NSAIDs to cause rebound headache (medication overuse headache), although less of a risk than ergotamine or triptans used for migraines.[5]
Toxicity
In humans, paracetamol has a narrow therapeutic index the therapeutic dose is close to the toxic dose. Additionally, paracetamol is contained in many preparations (both over-the-counter and prescription only medications). In some other animals, for example cats, all doses are toxic. This means that, despite being one of the safest analgesics available at recommended doses, there is a large potential for overdose and toxicity.[6] Without timely treatment, paracetamol overdose can lead to liver failure and death within days. Because of the wide over-the-counter availability of the drug, it is sometimes used in suicide attempts by those unaware of the prolonged timecourse, and high morbidity (likelihood of significant illness) and mortality associated with paracetamol-induced toxicity.
In the UK, sales of over-the-counter Paracetamol in pharmacies are restricted to packs of 32 tablets per customer per occasion (only 16 tablets in non-pharmacy stores). In Ireland, the limits are 24 and 12 tablets respectively.
Mechanism of toxicity
As discussed above in the section regarding metabolism, paracetamol is mostly converted to inactive compounds via Phase II metabolism by conjugation with sulfate and glucuronide, with a small portion being oxidized via the cytochrome P450 enzyme system. Cytochrome P450 2E1 (CYP2E1) converts paracetamol to a highly reactive intermediary metabolite, N-acetyl-p-benzo-quinone imine (NAPQI).
Under normal conditions, NAPQI is detoxified by conjugation with glutathione. In cases of paracetamol toxicity, the sulfate and glucuronide pathways become saturated, and more paracetamol is shunted to the cytochrome P450 system to produce NAPQI. As a result, hepatocellular supplies of glutathione become exhausted and NAPQI is free to react with cellular membrane molecules, resulting in widespread hepatocyte damage and death, clinically leading to acute hepatic necrosis. In animal studies, 70% of hepatic glutathione must be depleted before hepatotoxicity occurs.
Toxic dose
The toxic dose of paracetamol is highly variable. In adults, single doses above 10 grams or 150 mg/kg have a reasonable likelihood of causing toxicity.[7] Toxicity can also occur when multiple smaller doses within 24 hours exceeds these levels, or even with chronic ingestion of doses as low as 4 g/day, and death with as little as 6 g/day.
In children acute doses above 200 mg/kg could potentially cause toxicity. This higher threshold is largely due to children having relatively larger kidneys and livers than adults and hence being more tolerant of paracetamol overdose than adults.[8] Acute paracetamol overdose in children rarely causes illness or death with chronic supratherapeutic doses being the major cause of toxicity in children.
Since paracetamol is often included in combination with other drugs, it is important to include all sources of paracetamol when checking a person's dose for toxicity. In addition to being sold by itself, paracetamol may be included in the formulations of various analgesics and cold/flu remedies as a way to increase the pain-relieving properties of the medication. To prevent overdoses, one should read medication labels carefully for the presence of paracetamol and check with a pharmacist before using over-the-counter medications.
Risk factors for toxicity
Chronic excessive ethanol (alcohol) consumption can induce CYP2E1, thus increasing the potential toxicity of paracetamol.[9] For this reason, other analgesics such as aspirin or ibuprofen are sometimes recommended for hangovers.
Fasting is a risk factor, possibly because of depletion of hepatic glutathione reserves.
It is well documented that concomitant use of the CYP2E1 inducer isoniazid increases the risk of hepatotoxicity, though whether CYP2E1 induction is related to the hepatotoxicity in this case is unclear.[10][11] Concomitant use of other drugs which induce CYP enzymes such as antiepileptics (including carbamazepine, phenytoin, barbituates, etc) have also been reported as risk factors.
Natural history
Individuals who have overdosed on paracetamol generally have no specific symptoms for the first 24 hours. Although nausea, vomiting, and diaphoresis may occur initially, these symptoms generally resolve after several hours. After resolution of these symptoms, individuals tend to feel better, and may believe that the worst is over. If a toxic dose was absorbed, after this brief feeling of relative wellness, the individual develops overt hepatic failure. In massive overdoses, coma and metabolic acidosis may occur prior to hepatic failure.
Damage generally occurs in hepatocytes as they metabolize the paracetamol. Rarely, acute renal failure also may occur. This is usually caused by either hepatorenal syndrome or Multiple organ dysfunction syndrome. Acute renal failure may also be the primary clinical manifestation of toxicity. In these cases, it has been suggested that the toxic metabolite is produced more in the kidneys than in the liver.[12]
The prognosis of paracetamol toxicity varies depending on the dose and the appropriate treatment. In some cases, massive hepatic necrosis leads to fulminant hepatic failure with complications of bleeding, hypoglycemia, renal failure, hepatic encephalopathy, cerebral edema, sepsis, multiple organ failure, and death within days. In many cases, the hepatic necrosis may run its course, hepatic function may return, and the patient may survive with liver function returning to normal in a few weeks.
Diagnosis
Evidence of liver toxicity may develop in 1 to 4 days, although in severe cases it may be evident in 12 hours. Right upper quadrant tenderness may be present. Laboratory studies may show evidence of massive hepatic necrosis with elevated AST, ALT, bilirubin, and prolonged coagulation times (particularly, elevated prothrombin time). After paracetamol overdose, when AST and ALT exceed 1000 IU/L, paracetamol-induced hepatotoxicity can be diagnosed. However, the AST and ALT levels can exceed 10,000 IU/L. Generally the AST is somewhat higher than the ALT in paracetamol-induced hepatotoxicity.
A drug nomogram was developed in 1975 which estimated the risk of toxicity based on the serum concentration of paracetamol at a given number of hours after ingestion.[13] To determine the risk of potential hepatotoxicity, the paracetamol level is traced along the standard nomogram. A paracetamol level drawn in the first four hours after ingestion may underestimate the amount in the system because paracetamol may still be in the process of being absorbed from the gastrointestinal tract. Delay of the initial draw for the paracetamol level to account for this is not recommended since the history in these cases is often poor and a toxic level at any time is a reason to give the antidote.
Treatment
Initial measures
The initial treatment for uncomplicated paracetamol overdose, similar to any other overdose, is gastrointestinal decontamination. In addition, the antidote, acetylcysteine plays an important role. Paracetamol absorption from the gastrointestinal tract is complete within 2 hours under normal circumstances, so decontamination is most helpful if performed within this timeframe. Absorption may be somewhat slowed when it is ingested with food. There is considerable room for physician judgement regarding gastrointestinal decontamination, activated carbon administration is the most commonly used procedure, however, gastric lavage may also be considered if the amount ingested is potentially life threatening and the procedure can be performed within 60 minutes of ingestion.[14] Syrup of ipecac has no role in paracetamol overdose because the vomiting it induces delays the effective administration of activated carbon and oral acetylcysteine.
Activated carbon adsorbs paracetamol, reducing its gastrointestinal absorption. Administering activated carbon also poses less risk of aspiration than gastric lavage. Previously there was reluctance to give activated carbon in paracetamol overdose, because of concern that it may also absorb acetylcysteine. Studies have shown that no more than 39% of an oral acetylcysteine is absorbed when they are administered together.[15] Other studies have shown that activated carbon seems to be beneficial to the clinical outcome. It appears the most benefit from activated carbon is gained if it is given with 2 hours of ingestion.[16] However, administering activated carbon later than this can be considered in patients who may have delayed gastric emptying due to co-ingested drugs or following ingestion of sustained or delayed release paracetamol preparations. Activated carbon should also be administered if co-ingested drugs warrant decontamination. There are conflicting recommendations[15][17] regarding whether to change the dosing of oral acetylcysteine after the administration of activated carbon, and even whether the dosing of acetylcysteine needs to be altered at all.
Acetylcysteine
Acetylcysteine works to reduce paracetamol toxicity by supplying sulfhydryl groups (mainly in the form of glutathione, of which it is a precursor) to react with the toxic NAPQI metabolite so that it does not damage cells and can be safely excreted.
If the patient presents less than 8 hours after paracetamol overdose, then acetylcysteine significantly reduces the risk of serious hepatotoxicity. If NAC is started more than 8 hours after ingestion, there is a sharp decline in its effectiveness because the cascade of toxic events in the liver has already begun and the risk of acute hepatic necrosis and death increases dramatically. Although acetylcysteine is most effective if given early, it still has beneficial effects if given as late as 48 hours after ingestion.[18] In clinical practice, if the patient presents more than 8 hours after the paracetamol overdose, then activated charcoal is probably not useful, and acetylcysteine is started immediately. In earlier presentations the doctor can give charcoal as soon as the patient arrives, start giving acetylcysteine, and wait for the paracetamol level from the laboratory.
In United States practice, intravenous (IV) and oral administration are considered to be equally effective. However, IV is the only recommended route in Australasian and British practice.
Oral acetylcysteine is given as a 140 mg/kg loading dose followed by 70 mg/kg every 4 hours for 17 more doses. Oral acetylcysteine may be poorly tolerated due to its unpleasant taste, odor, and its tendency to cause nausea and vomiting. It can be diluted to a 5% solution, from its marketed 10% or 20% solutions, to improve palatability. Where oral acetylcysteine is required, the inhalation formulation of acetylcysteine (Mucomyst) is often given orally. The respiratory formulation can also be diluted and filter sterilized by a hospital pharmacist for IV use, however this is an uncommon practice. If repeat doses of charcoal are indicated because of another ingested drug, then subsequent doses of carbon and acetylcysteine should be staggered every two hours.
Intravenous acetylcysteine (Parvolex/Acetadote) is used as a continuous intravenous infusion over 20 hours (total dose 300 mg/kg). Recommended administration involves infusion of a 150 mg/kg loading dose over 15 minutes, followed by 50 mg/kg infusion over 4 hours; the last 100 mg/kg are infused over the remaining 16 hours of the protocol. Intravenous acetylcysteine has the advantage of shortening hospital stay, increasing both doctor and patient convenience, and it allows administration of activated carbon to reduce absorption of both the paracetamol and any co-ingested drugs without concerns about interference with oral acetylcysteine.[19]
Baseline laboratory studies include bilirubin, AST, ALT, and prothrombin time (with INR). Studies are repeated at least daily. Once it has been determined that a potentially toxic overdose has occurred, acetylcysteine is continued for the entire regimen, even after the paracetamol level becomes undetectable in the blood. If hepatic failure develops, acetylcysteine should be continued beyond the standard doses until hepatic function improves or until the patient has a liver transplant.
Prognosis
The mortality rate from paracetamol overdose increases 2 days after the ingestion, reaches a maximum on day 4, and then gradually decreases. Patients with a poor prognosis are usually identified for likely liver transplantation. Acidemia is the most important single indicator of probable mortality and the need for transplantation. A mortality rate of 95% without transplant was reported in patients who had a documented pH of < 7.30. Other indicators of poor prognosis include renal insufficiency, grade 3 or worse hepatic encephalopathy, a markedly elevated prothrombin time, or a rise in prothrombin time from day 3 to day 4. One study has shown that a factor V level less than 10% of normal indicated a poor prognosis (91% mortality) while a ratio of factor VIII to factor V of less than 30 indicated a good prognosis (100% survival).
Danger to animals
Paracetamol is extremely toxic to cats, and should not be given to them under any circumstances. Cats lack the necessary glucuronyl transferase enzymes to safely break paracetamol down and tiny fractions of a normal tablet for humans may prove fatal.[20]
In dogs paracetamol is a useful anti-inflammatory with a good safety record, causing a lower incidence of gastric ulceration than NSAIDs. It should only be administered on veterinary advice. A paracetamol codene product (Pardale-V)[2] licensed for use in dogs is available on veterinary prescription in the UK.
Any cases of suspected ingestion in cats or overdose in dogs should be taken to a veterinarian immediately for detoxification.[21] The effects of toxicity can include liver damage, haemolytic anaemia, oxidative damage to the red blood cells and bleeding tendencies. There are no home remedies, and the amount of irreversible liver failure is dependent on how quickly veterinary intervention begins. Treatment of paracetamol overdose by a veterinarian may involve the use of supportive fluid therapy, acetylcysteine (Mucomyst), methionine or s-adenosyl-l-methionine (SAMe) to slow liver damage and cimetidine (Tagamet) to protect against gastric ulceration. Once liver damage has occurred it cannot be reversed.[3]
November 2006 United States recall of generic paracetamol
On November 9, 2006, the U.S. Food and Drug Administration (FDA) announced a recall of approximately 11 million bottles of generic paracetamol manufactured by Perrigo, the largest manufacturer of generic over-the-counter drugs in the United States, due to metal particles having been found in a small sample of paracetamol caplets.[22] The recall is limited to 383 lots of 500 mg caplets; other lots and other dosages, as well as generic paracetamol from other manufacturers or proprietary formulations of paracetamol, are not affected. The complete list of recalled lots may be found on the FDA website.
As of November 14, 2006, no deaths or injuries have been reported as a result of the incident.
References
- ^ Brodie BB, Axelrod J (1948). "The fate of acetanilide in man". J Pharmacol Exp Ther 94 (1): 29-38.
- ^ Boutaud O, Aronoff DM, Richardson JH, Marnett LJ, Oates JA (2002). "Determinants of the cellular specificity of acetaminophen as an inhibitor of prostaglandin H2 synthases". Proc Natl Acad Sci U S A 99 (10): 7130-5. PMID 12011469.
- ^ Swierkosz TA, Jordan L, McBride M, McGough K, Devlin J, Botting RM (2002). "Actions of paracetamol on cyclooxygenases in tissue and cell homogenates of mouse and rabbit" (PDF). Med Sci Monit 8 (12): BR496-503. PMID 12503027.
- ^ Pina LA, Sandrini M, Vitale G (July 11, 1996). "The antinociceptive action of paracetamol is associated with changes in the serotonergic system in the rat brain". Eur J Pharmacol 308 (1): 31-40. DOI:10.1016/0014-2999(96)00261-0. PMID 8836629. Retrieved on 2006-04-25.
- ^ Colás Chacartegui R, Temprano González R, Gómez Arruza C, Muñoz Cacho P, Pascual Gómez J (2005). "[Abuse pattern of analgesics in chronic daily headache: a study in the general population]". Rev Clin Esp 205 (12): 583-7. PMID 16527179.
- ^ Sheen C, Dillon J, Bateman D, Simpson K, Macdonald T (2002). "Paracetamol toxicity: epidemiology, prevention and costs to the health-care system.". QJM 95 (9): 609-19. PMID 12205339.
- ^ Dart RC, Erdman AR, Olson KR, Christianson G, Manoguerra AS, Chyka PA, Caravati EM, Wax PM, Keyes DC, Woolf AD, Scharman EJ, Booze LL, Troutman WG; American Association of Poison Control Centers. (2006). "Acetaminophen poisoning: an evidence-based consensus guideline for out-of- hospital management.". Clin Toxicol (Phila) 44 (1): 1-18. PMID 16496488.
- ^ Tenenbein M (2004). "Acetaminophen: the 150 mg/kg myth.". J Toxicol Clin Toxicol 42 (2): 145-8. PMID 15214618.
- ^ Zimmerman HJ, Maddrey WC (1995). "Acetaminophen (paracetamol) hepatotoxicity with regular intake of alcohol: analysis of instances of therapeutic misadventure". Hepatology 22 (3): 767-73. PMID 7657281.
- ^ Crippin JS (1993). "Acetaminophen hepatotoxicity: potentiation by isoniazid". Am J Gastroenterol 88 (4): 590-2. PMID 8470644.
- ^ Nolan CM, Sandblom RE, Thummel KE, Slattery JT, Nelson SD (1994). "Hepatotoxicity associated with acetaminophen usage in patients receiving multiple drug therapy for tuberculosis". Chest 105 (2): 408-11. PMID 7508362.
- ^ Boutis K, Shannon M (2001). "Nephrotoxicity after acute severe acetaminophen poisoning in adolescents". J Toxicol Clin Toxicol 39 (5): 441-5. PMID 11545233.
- ^ Rumack B, Matthew H (1975). "Acetaminophen poisoning and toxicity". Pediatrics 55 (6): 871-6. PMID 1134886.
- ^ Vale JA, Kulig K; American Academy of Clinical Toxicology; European Association of Poisons Centres and Clinical Toxicologists. (2004). "Position paper: gastric lavage". J Toxicol Clin Toxicol 42 (7): 933-43. PMID 15641639.
- ^ a b Ekins B, Ford D, Thompson M, Bridges R, Rollins D, Jenkins R (1987). "The effect of activated charcoal on N-acetylcysteine absorption in normal subjects". Am J Emerg Med 5 (6): 483-7. PMID 3663288.
- ^ Buckley NA, Whyte IM, O'Connell DL, Dawson AH. (1999). "Activated charcoal reduces the need for N-acetylcysteine treatment after acetaminophen (paracetamol) overdose". J Toxicol Clin Toxicol 37 (6): 753-7. PMID 10584587.
- ^ Spiller H, Krenzelok E, Grande G, Safir E, Diamond J (1994). "A prospective evaluation of the effect of activated charcoal before oral N-acetylcysteine in acetaminophen overdose". Ann Emerg Med 23 (3): 519-23. PMID 8135427.
- ^ Keays R, Harrison P, Wendon J, Forbes A, Gove C, Alexander G, Williams R (1991). "Intravenous acetylcysteine in paracetamol induced fulminant hepatic failure: a prospective controlled trial". BMJ 303 (6809): 1026-9. PMID 1954453.
- ^ Buckley N, Whyte I, O'Connell D, Dawson A (1999). "Oral or intravenous N-acetylcysteine: which is the treatment of choice for acetaminophen (paracetamol) poisoning?". J Toxicol Clin Toxicol 37 (6): 759-67. PMID 10584588.
- ^ Allen AL (2003). "The diagnosis of acetaminophen toxicosis in a cat". Can Vet J 44 (6): 509-10. PMID 12839249.
- ^ Villar D, Buck WB, Gonzalez JM (1998). "Ibuprofen, aspirin and acetaminophen toxicosis and treatment in dogs and cats". Vet Hum Toxicol 40 (3): 156-62. PMID 9610496.
- ^ U.S. Food and Drug Administration (November 9, 2006). FDA Informs Public of Nationwide Recall of 500mg Strength Store-Brand Acetaminophen Caplets. Press release. Retrieved on 2006-11-14.
